Working with Affymetrix Data
This example shows how to use the functions in the Bioinformatics Toolbox™ for working with Affymetrix® GeneChip® data.
About Affymetrix Data Files
The function affyread can read four types of Affymetrix data files. These are DAT files, which contain raw image data, CEL files which contain information about the intensity values of the individual probes, CHP files which contain information about probe sets, and EXP files, which contain information about experimental conditions and protocols. affyread can also read CDF and GIN library files. The CDF file contains information about which probes belong to which probe set and the GIN file contains information about the probe sets such as the gene name with which the probe set is associated. Most of the data sets are stored in DTT archives. To extract the DAT, CEL and CHP files you need to install the Data Transfer Tool.
Downloading the E. coli Antisense Data Set
You need some sample data files (DAT, CEL, CHP) from here. This example uses the E. coli Antisense Genome Array. Extract the data files using the Data Transfer Tool. Set the variable exampleDataDir to the name of the path and directory to which you extracted the sample data files.
exampleDataDir = 'C:\Examples\affydemo\data';
Downloading E. coli Antisense Library Files
In addition to the data files, you also need Ecoli_ASv2.CDF and Ecoli_ASv2.GIN, the library files for the E. coli Antisense Genome Array. You may already have these files if you have any Affymetrix GeneChip software installed on your machine. If not, get the library files by downloading the E. coli Antisense Genome Array zip file from here.
Note that you will have to register in order to access the library files.
You only have to unzip the files, you do not have to run the Setup.exe file in the archive.
Set the variable libDir to the name of the path and directory to which you extracted the library files.
libDir = 'C:\Examples\affydemo\libfiles';
Image Files (DAT Files)
The raw image data from the chip scanner is saved in the DAT file. If you use affyread to read a DAT file you will see that it creates a MATLAB® structure.
datStruct = affyread(fullfile(exampleDataDir,'Ecoli-antisense-121502.dat'));
You can access fields of the structure using the dot notation.
datStruct.NumRows
ans =
4733
Displaying an Image File
You can use the imagesc command to display the image.
datFigure = figure; imagesc(datStruct.Image);
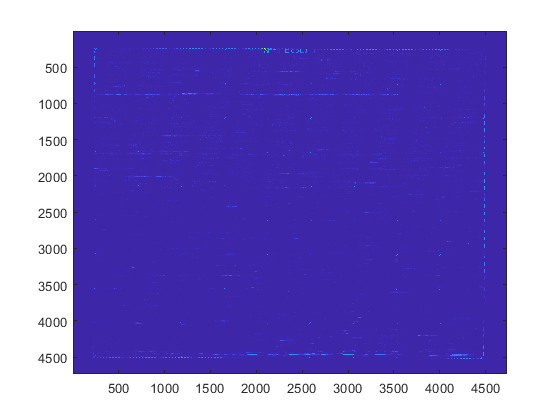
You can change the colormap from the default jet to another using the colormap command.
colormap pink
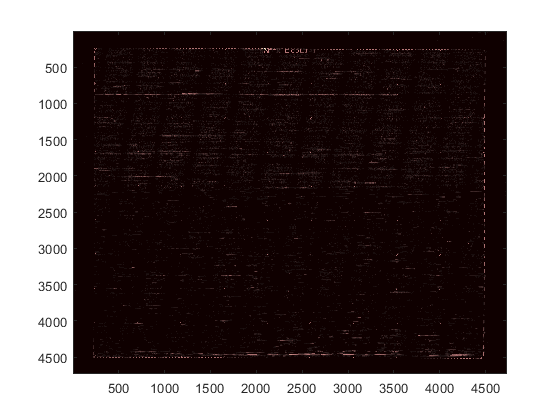
You can zoom in on a particular area by using the Zoom In tool with the mouse, or by using the axis command. Notice that this stretches the y-axis.
axis([1900 2800 160 650])
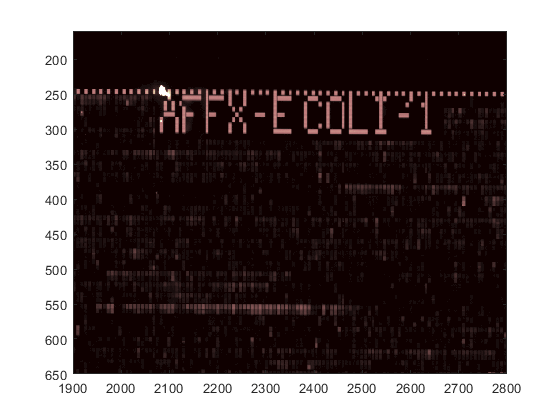
You can use the axis image command to set the correct aspect ratio.
axis image
axis([1900 2800 160 650])
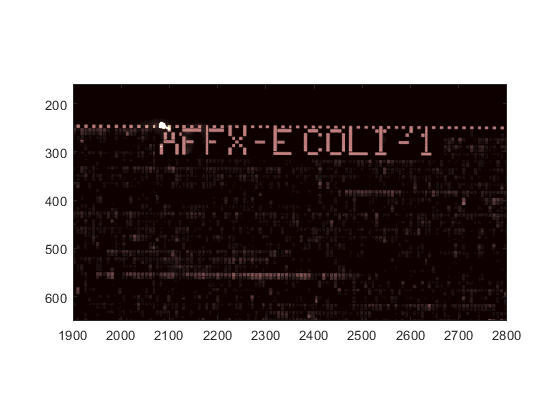
Probe Results Files (CEL Files)
The information about each probe on the chip is extracted from the image data by the Affymetrix image analysis software. The information is stored in the CEL file. affyread reads a CEL file into a structure. Notice that many of the fields are the same as those in the DAT structure.
celStruct = affyread(fullfile(exampleDataDir,'Ecoli-antisense-121502.CEL'));
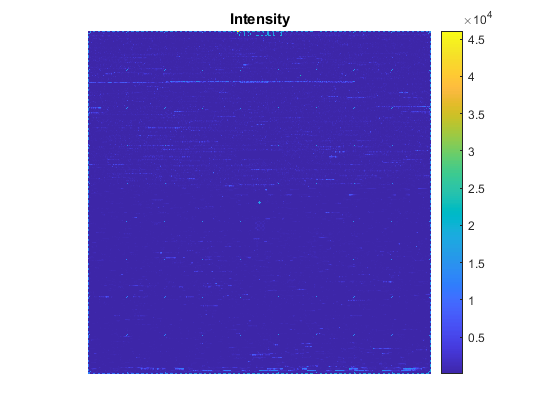
The CEL file contains information about where each probe is on the chip and also the intensity values for the probe. You can use the maimage function to display the chip.
celFigure = figure; maimage(celStruct)
Again, you can zoom in on a specific region.
axis([200 340 0 70])
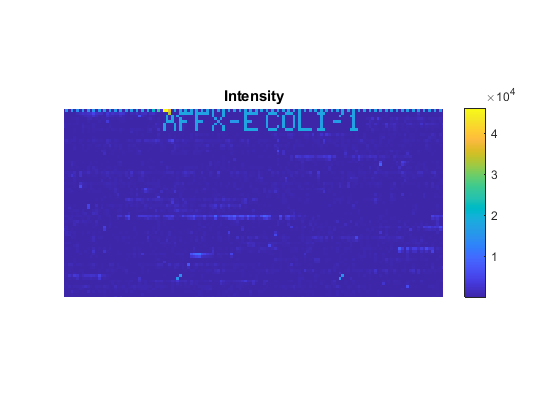
If you compare the image created from the CEL file and the image created from the DAT file, you will notice that the CEL image is lower resolution. This is because there is only one pixel per probe in this image, whereas the DAT file image has many pixels per probe.
The structures created by affyread can be very large. It is a good idea to clear them from memory once they are no longer needed.
clear datStruct
close(datFigure); close(celFigure);
The Probes field of the CEL structure contains information about the individual probes. There are eight values per probe. These are stored in the ProbeColumnNames field of the structure.
celStruct.ProbeColumnNames
ans =
8×1 cell array
{'PosX' }
{'PosY' }
{'Intensity'}
{'StdDev' }
{'Pixels' }
{'Outlier' }
{'Masked' }
{'ProbeType'}
So if you look at one row of the Probes field of the CEL structure you will see eight values corresponding to the X position, Y position, intensity, and so forth.
celStruct.Probes(1:10,:)
ans =
10×8 single matrix
1.0e+04 *
Columns 1 through 7
0 0 0.0082 0.0030 0.0036 0 0
0.0001 0 1.4202 0.3160 0.0036 0 0
0.0002 0 0.0080 0.0014 0.0030 0 0
0.0003 0 1.4760 0.2265 0.0036 0 0
0.0004 0 0.0050 0.0014 0.0036 0 0
0.0005 0 0.0073 0.0015 0.0036 0 0
0.0006 0 1.3595 0.2367 0.0036 0 0
0.0007 0 0.0087 0.0018 0.0036 0 0
0.0008 0 1.3284 0.2926 0.0036 0 0
0.0009 0 0.0104 0.0018 0.0030 0 0
Column 8
0.0001
0.0001
0.0001
0.0001
0.0001
0.0001
0.0001
0.0001
0.0001
0.0001
Results Files (CHP Files)
The CHP file contains the results of the experiment. These include the average signal measures for each probe set as determined by the Affymetrix software and information about which probe sets are called as present, absent or marginal and the p-values for these calls.
chpStruct = affyread(fullfile(exampleDataDir,'Ecoli-antisense-121502.CHP'),libDir);
The ProbeSets field contains information about the probe sets. This includes some library information, such as the ID and the type of probe set, and also results information such as the calculated signal value and the Present/Absent/Marginal call information. The call is given in the Detection field of the ProbeSets structure. The 'argG_b3172_at' probe set is called as being 'Present'.
chpStruct.ProbeSets(5213)
ans =
struct with fields:
Name: 'argG_b3172_at'
ProbeSetType: 'Expression'
CompDataExists: 0
NumPairs: 15
NumPairsUsed: 15
Signal: 127.6070
Detection: 'Present'
DetectionPValue: 0.0134
CommonPairs: []
SignalLogRatio: []
SignalLogRatioLow: []
SignalLogRatioHigh: []
Change: []
ChangePValue: []
However, the 'IG_2069_3319273_3319712_rev_at' probe set is called 'Absent'.
chpStruct.ProbeSets(5216)
ans =
struct with fields:
Name: 'IG_2069_3319273_3319712_rev_at'
ProbeSetType: 'Expression'
CompDataExists: 0
NumPairs: 15
NumPairsUsed: 15
Signal: 35.0037
Detection: 'Absent'
DetectionPValue: 0.2661
CommonPairs: []
SignalLogRatio: []
SignalLogRatioLow: []
SignalLogRatioHigh: []
Change: []
ChangePValue: []
And the 'yhbX_b3173_at' probe set is called 'Marginal'.
chpStruct.ProbeSets(5215)
ans =
struct with fields:
Name: 'yhbX_b3173_at'
ProbeSetType: 'Expression'
CompDataExists: 0
NumPairs: 15
NumPairsUsed: 15
Signal: 147.7237
Detection: 'Marginal'
DetectionPValue: 0.0559
CommonPairs: []
SignalLogRatio: []
SignalLogRatioLow: []
SignalLogRatioHigh: []
Change: []
ChangePValue: []
You can calculate how many probe sets are called as being 'Present',
numPresent = sum(strcmp('Present',{chpStruct.ProbeSets.Detection}))
numPresent =
4605
'Absent',
numAbsent = sum(strcmp('Absent',{chpStruct.ProbeSets.Detection}))
numAbsent =
2524
and 'Marginal'.
numMarginal = sum(strcmp('Marginal',{chpStruct.ProbeSets.Detection}))
numMarginal = 183
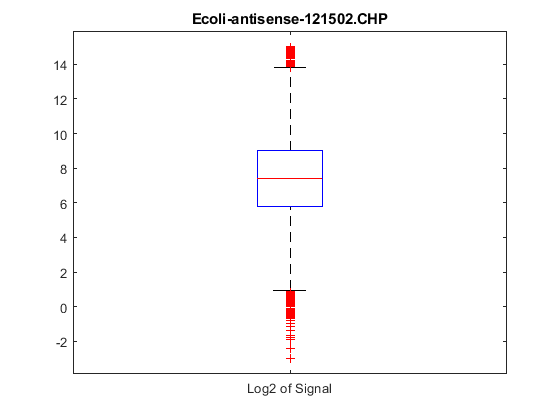
maboxplot will display a box plot of the log2 signal values for all probe sets.
maboxplot(chpStruct,'Signal','title',chpStruct.Name)
Library Files (CDF Files)
The CHP file gives summary information about probe sets but if you want more detailed information about how the individual probes in a probe set behave you need to connect the probe information in the CEL file to the corresponding probe sets. This information is stored in the CDF library file associated with a chip type. The CDF files are typically stored in a central library directory.
cdfStruct = affyread('Ecoli_ASv2.cdf',libDir);
Most of the information in the file is about the probe sets. In this example there are 7312 regular probe sets and 13 QC probe sets. The ProbeSets field of the structure is a 7325x1 array of structures.
cdfStruct.ProbeSets
ans =
7325×1 struct array with fields:
Name
ProbeSetType
CompDataExists
NumPairs
NumQCProbes
QCType
GroupNames
ProbePairs
A probe set record contains information about the name, type and number of probe pairs in the probe set.
probeSetIndex = 5213; cdfStruct.ProbeSets(probeSetIndex)
ans =
struct with fields:
Name: 'argG_b3172_at'
ProbeSetType: 'Expression'
CompDataExists: 0
NumPairs: 15
NumQCProbes: 0
QCType: 0
GroupNames: {'argG_b3172_at'}
ProbePairs: [15×6 int32]
The information about where the probes for a probe set are on the chip is stored in the ProbePairs field. This is a matrix with one row for each probe pair and six columns. The information in the columns corresponds to the ProbeSetColumnNames of the CDF structure.
cdfStruct.ProbeSetColumnNames cdfStruct.ProbeSets(probeSetIndex).ProbePairs
ans =
6×1 cell array
{'GroupNumber'}
{'Direction' }
{'PMPosX' }
{'PMPosY' }
{'MMPosX' }
{'MMPosY' }
ans =
15×6 int32 matrix
1 2 430 177 430 178
1 2 431 177 431 178
1 2 432 177 432 178
1 2 433 177 433 178
1 2 434 177 434 178
1 2 435 177 435 178
1 2 436 177 436 178
1 2 437 177 437 178
1 2 438 177 438 178
1 2 439 177 439 178
1 2 440 177 440 178
1 2 441 177 441 178
1 2 442 177 442 178
1 2 443 177 443 178
1 2 444 177 444 178
The first column shows the probe group number. The second column shows the probe direction. The group number is always 1 for expression arrays. Direction 1 corresponds to 'sense' and 2 corresponds to 'anti-sense'. The remaining columns give the X and Y coordinates of the PM and MM probes on the chip. You can use these coordinates to find the index of a probe in the celStruct.
PMX = cdfStruct.ProbeSets(probeSetIndex).ProbePairs(1,3);
PMY = cdfStruct.ProbeSets(probeSetIndex).ProbePairs(1,4);
theProbe = find((celStruct.Probes(:,1) == PMX) & ...
(celStruct.Probes(:,2) == PMY))
theProbe =
96719
You can then extract all the information about this probe from the CEL structure.
celStruct.Probes(theProbe,:)
ans =
1×8 single row vector
Columns 1 through 7
430.0000 177.0000 169.0000 35.4000 25.0000 0 0
Column 8
1.0000
If you want to do this lookup for all probes, you can use the function probelibraryinfo. This creates a matrix with one row per probe and three columns. The first column is the index of the probe set to which the probe belongs. The second column contains the probe pair index and the third column indicates if the probe is a perfect match (1) or mismatch (-1) probe. Notice that index of the probe pair index is 1 based.
probeinfo = probelibraryinfo(celStruct,cdfStruct); probeinfo(theProbe,:)
ans =
5213 1 1
The function probesetvalues does the reverse of this lookup and creates a matrix of information from the CEL and CDF structures containing all the information about a given probe set. This matrix has 20 columns corresponding to ProbeSetNumber, ProbePairNumber, UseProbePair, Background, PMPosX, PMPosY, PMIntensity, PMStdDev, PMPixels, PMOutlier, PMMasked, MMPosX, MMPosY, MMIntensity, MMStdDev, MMPixels, MMOutlier, MMMasked, Group, and Direction.
probeName = cdfStruct.ProbeSets(probeSetIndex).Name; psvals = probesetvalues(celStruct,cdfStruct,probeName); sprintf( ['%4d %2d %d %d PM: %3d %3d %5.1f %5.1f %2d %d %d',... ' MM: %3d %3d %5.1f %5.1f %2d %d %d %d %d\n'],psvals')
ans =
'5212 0 0 4.543512e+01 PM: 430 177 169.0 35.4 25 0 0 MM: 430 178 163.5 24.1 30 0 0 1 2
5212 1 0 4.545356e+01 PM: 431 177 127.3 21.8 30 0 0 MM: 431 178 100.3 14.6 36 0 0 1 2
5212 2 0 4.547230e+01 PM: 432 177 127.0 23.7 30 0 0 MM: 432 178 175.0 28.6 36 0 0 1 2
5212 3 0 4.549129e+01 PM: 433 177 133.3 25.9 36 0 0 MM: 433 178 94.0 22.7 30 0 0 1 2
5212 4 0 4.551051e+01 PM: 434 177 212.3 43.3 36 0 0 MM: 434 178 171.8 36.5 30 0 0 1 2
5212 5 0 4.552995e+01 PM: 435 177 149.5 27.5 36 0 0 MM: 435 178 154.0 30.3 30 0 0 1 2
5212 6 0 4.554958e+01 PM: 436 177 50.3 11.2 30 0 0 MM: 436 178 46.0 9.8 25 0 0 1 2
5212 7 0 4.556938e+01 PM: 437 177 152.5 37.7 36 0 0 MM: 437 178 107.0 21.0 36 0 0 1 2
5212 8 0 4.558934e+01 PM: 438 177 164.5 31.2 36 0 0 MM: 438 178 97.3 21.9 36 0 0 1 2
5212 9 0 4.560939e+01 PM: 439 177 126.0 23.4 36 0 0 MM: 439 178 121.3 25.3 36 0 0 1 2
5212 10 0 4.562955e+01 PM: 440 177 54.0 11.2 36 0 0 MM: 440 178 54.0 12.9 36 0 0 1 2
5212 11 0 4.564975e+01 PM: 441 177 83.3 17.4 36 0 0 MM: 441 178 62.3 12.5 36 0 0 1 2
5212 12 0 4.566998e+01 PM: 442 177 95.5 17.1 30 0 0 MM: 442 178 84.0 18.6 30 0 0 1 2
5212 13 0 4.569022e+01 PM: 443 177 110.0 19.6 36 0 0 MM: 443 178 92.5 22.0 36 0 0 1 2
5212 14 0 4.571042e+01 PM: 444 177 251.0 46.0 36 0 0 MM: 444 178 111.8 20.7 36 0 0 1 2
'
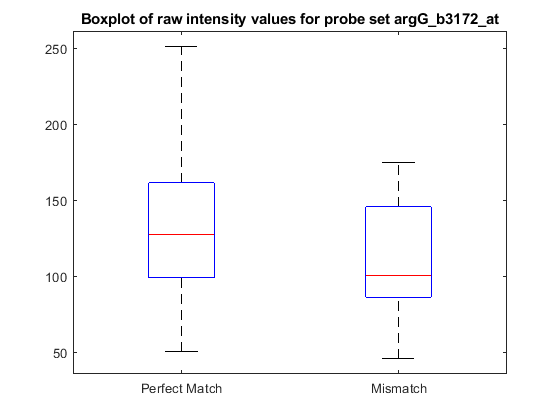
You can extract the intensity values from the matrix and look at some of the statistics of the data.
pmIntensity = psvals(:,7); mmIntensity = psvals(:,14); boxplot([pmIntensity,mmIntensity],'labels',{'Perfect Match','Mismatch'}) title(sprintf('Boxplot of raw intensity values for probe set %s',... probeName),'interpreter','none') % Use interpreter none to prevent the TeX interpreter treating the _ as % subscript.
Plotting the Probe Set Values
Now that you have the intensity values for the probes, you can plot the values for the perfect match and mismatch probes.
figure plot(pmIntensity,'b'); hold on plot(mmIntensity,'r'); hold off title(sprintf('Probe intensity values for probe set %s',... probeName),'interpreter','none')
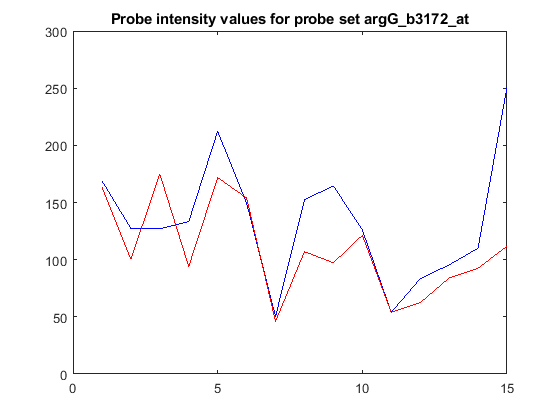
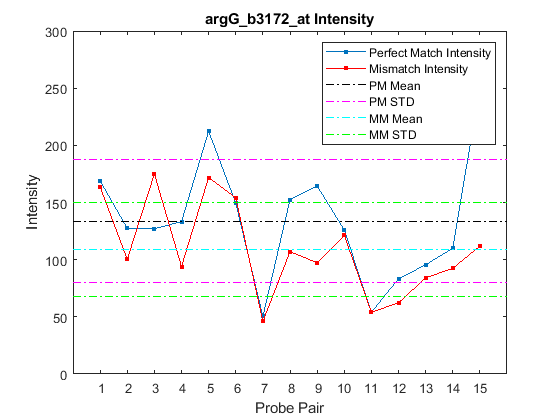
Alternatively, you can use the function probesetplot to create this plot directly from the CEL and CDF structures. The showstats option adds the mean, and lines for +/- one standard deviation for both the perfect match and the mismatch probes to the plot.
probesetplot(celStruct,cdfStruct,probeName,'showstats',true);
Gene Names and Probe Set IDs
The Affymetrix probe set IDs are not particularly descriptive. The mapping between the probe set IDs and the gene IDs is stored in the GIN library file. This is a text file so you can open it in an editor and browse through the file, or you can use affyread to read the information into a structure.
ginStruct = affyread('Ecoli_ASv2.GIN',libDir)
ginStruct =
struct with fields:
Name: 'Ecoli_ASv2'
Version: 2
ProbeSetName: {7312×1 cell}
ID: {7312×1 cell}
Description: {7312×1 cell}
SourceNames: {2×1 cell}
SourceURL: {2×1 cell}
SourceID: [7312×1 double]
You can search through the structure for a particular probe set. Alternatively, you can use the function probesetlookup to find information about the gene for a probe set.
info = probesetlookup(cdfStruct,probeName)
info =
struct with fields:
Identifier: '3315278'
ProbeSetName: 'argG_b3172_at'
CDFIndex: 5213
GINIndex: 3074
Description: '/start=3316278 /end=3317621 /direction=+ /description=argininosuccinate synthetase'
Source: 'NCBI EColi Genome'
SourceURL: 'http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/altvik?gi=115&db=g&from=3315278'
Affymetrix, GeneChip, and NetAffx are registered trademarks of Affymetrix, Inc.
You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)